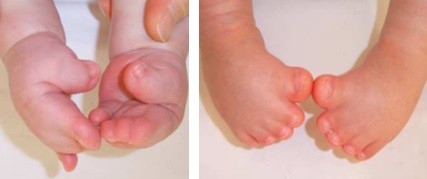
Síndrome descrita por Rudolf Pfeiffer em 1964, ela apresenta uma braquicefalia (fechamento das suturas cranianas coronais) associada a sindactilias membranosas variáveis das mãos e pés e sobretudo um alargamento dos polegares e dos dedões dos pés (“dedão do pé aumentado”), com um desvio marcante para dentro. Uma braquidactilia, uma estenose dos cotovelos, e até mesmo sinfalangia são frequentementes descritos.
As formas graves, com o crânio em trevo, são descritas. Nestes casos, a dismorfia craniana é muito importante, devido a um bombeamento considerável das fossas temporais e de uma estenose lateral das regiões frontoparietais, levando a um aspecto trilobado visto de frente. A hidrocefalia congênita é constante. Essa dismorfia maior, muitas vezes chamada síndrome do crânio em trevo, pode igualmente ser observada nas síndromes de Apert e na forma precoce da síndrome de Crouzon.
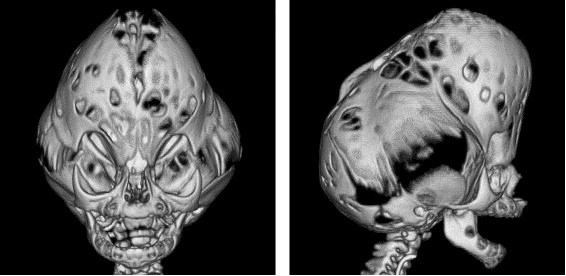
A Síndrome de Pfeiffer é rara, com uma incidência aproximada de 1 para cada 100.000 indivíduos. Ocorre cranioestenoses em associação com polegares e dedões dos pés curtos e desviados para dentro. A fusão prematura das suturas coronais e lambdóides pode ser acompanhada ocasionalmente da fusão ainda da sutura sagital, levando à forma anormal do crânio. Ocorre uma aparência facial característica, com alargamento da cabeça e achatamento do occipital, alongamento da região frontal, hipoplasia (menor desenvolvimento) do terço médio da face, nariz curto com dorso nasal baixo e afastamento dos olhos (hipertelorismo ocular). Os pacientes geralmente apresentam olhos proeminentes (proptose ocular) devido às órbitas serem muito rasas.
Os polegares e os dedões dos pés são curtos e tortos, com um desvio típico. Ocorre junção (sindactilia) do segundo e terceiro dedos. Anomalias adicionais podem incluir: retardo mental em graus variados, estenose de aqueduto cerebral com consequente hidrocefalia, implantação baixa das orelhas, estenose (extreitamento) do conduto auditivo externo, infecções de ouvido recorrentes, e menos frequentemente, hidronefrose, rins pélvicos, bexiga pouco desenvolvida (hipoplásica).
Pacientes com Síndrome de Pfeiffer podem apresentar obstrução de vias aéreas superiores devido à retrusão do terço médio da face.
De acordo com as características clínicas, a Síndrome de Pfeiffer pode ser dividida em 3 subtipos:
Tipo 1: Síndrome de Pfeiffer “clássica”, com manifestações leves incluindo braquicefalia, hipoplasia do terço médio da face e anomalias de dedos das mãos e dos pés. Está associada com desenvolvimento neurológico e intelectual normais e geralmente tem um bom prognóstico.
Tipo 2: caracteriza-se pela deformidade do crânio trilobado, com uma proeminência no topo e uma em cada lado da cabeça, dando um aspecto em forma de “ folha de trevo” (também chamado de Kleeblattschädel), embora essa característica não seja exclusiva da Síndrome de Pfeiffer, podendo ocorrer em outras síndromes ou mesmo isoladamente. Apresenta ainda proptose (olhos saltados) grave, anomalias de dedos das mãos e pés, anquilose (junção) ou estenose de cotovelo, atraso de desenvolvimento mental e complicações neurológicas. O crânio em forma de trevo pode causar limitação ao crescimento cerebral e a proptose grave pode causar perdas visuais importantes.
Tipo 3: semelhante ao tipo 2, porém sem deformidade do crânio em forma de trevo. A ausência do crânio em forma de trevo pode tornar difícil estabelecer o diagnóstico. Os tipos 2 e 3 ocorrem esporadicamente e apresentam um risco aumentado de morte precoce devido ao comprometimento neurológico grave e problemas respiratórios. Uma superposição clínica entre os três tipos pode ocorrer.
Diagnosticando a Síndrome de Pfeiffer
O diagnóstico é geralmente clínico, baseado na presença de cranioestenoses e polegares e dedões dos pés alterados. Geneticamente, ocorrem mutações nos genes dos receptores dos fatores de crescimento dos fibroblastos (do inglês: fibroblast growth factor receptor – FGFR1 e FGFR2). Esses genes atuam na sinalização das células em resposta ao seu ambiente, regulando processos de proliferação, diferenciação e migração celular. Uma mutação em um desses genes causa uma sinalização prolongada, que pode provocar a maturação precoce das células ósseas e fusão prematura dos ossos do crânio, das mãos e dos pés.
Faz diagnóstico diferencial com outras síndromes de craniofacioestenoses, como as Síndromes de Crouzon e Apert, discutidas em outro tópico. De fato, as semelhanças clínicas são grandes, principalmente no que diz respeito ao aspecto orbitário (órbitas rasas com proptose ocular) e à retrusão do terço médio da face, embora geneticamente sejam distintas.


