Outras Malformações
Citando os tipos específicos de malformações, ajudando na compreensão do tema.
O que são?
As fissuras de lábio e palato, conhecidas também como lábio leporino (termo em desuso) e fenda palatina (ou “céu da boca aberto”), correspondem a malformação congênita mais comum da face, com uma incidência aproximada de 1 caso para cada 700 nascimentos. A criança pode apresentar apenas uma fissura de lábio, uma fissura de lábio e palato ou apenas a fissura do palato. A fissura do lábio pode ser incompleta ou completa, com comprometimento unilateral ou bilateral. A fissura do palato também pode ter extensão variável e ser unilateral ou bilateral.
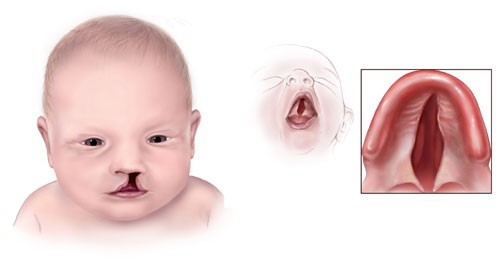
Causas de fissura de lábio e fenda palatina
Em cerca de um terço dos casos, a criança com fissura apresenta um parente que também teve fissura. Nos outros dois terços dos casos, não há nenhuma história familiar. A fissura não acontece em consequência de algum fato específico ocorrido com a mãe durante a gestação. Foram descobertos diversos genes associados à fissura labiopalatina, e várias drogas diferentes (álcool, cigarros, algumas medicações anticonvulsivantes, derivados de vitamina A) também estão relacionadas a uma maior incidência de fissuras. No entanto, o mais provável é que diversos fatores contribuam para a ocorrência de uma fissura, caracterizando uma origem multifatorial.
Com o avanço da ultrassonografia, muitas fissuras podem ser diagnosticadas intraútero. Apesar disso, algumas crianças têm seu diagnóstico realizado apenas ao nascimento, causando surpresa para os médicos e principalmente para os familiares. Algumas fissuras de palato podem ter um diagnóstico ainda mais tardio, quando a criança inicia a fala. São as fissuras de palato submucosas ou fissuras ocultas, em que a mucosa oral se encontra íntegra, mas a musculatura e os ossos estão separados. Crianças nascidas com fissura de lábio e palato devem ser avaliadas por médicos experientes, investigando e afastando outros problemas que podem estar associados ao quadro. As crianças que apresentam uma fissura isolada do palato, especialmente se elas também apresentam uma mandíbula pequena, devem ser avaliadas por um especialista em genética médica para afastar o diagnóstico de Sequência de Pierre Robin, Síndrome Velocardiofacial, dentre outras síndromes.
O tratamento a ser empregado vai depender do tipo de fissura que a criança apresentar, seja uma fissura isolada de lábio, seja uma fissura lábiopalatina ou uma fissura isolada de palato. Existem diversas técnicas descritas na literatura para o tratamento das fissuras de lábio e palato e os cirurgiões podem divergir em relação à técnica a ser utilizada, bem como a idade adequada para o reparo. É necessário tirar todas as suas dúvidas em relação à cronologia e ao tipo de tratamento que vai ser empregado pelo seu cirurgião. Algumas questões básicas relacionadas ao tratamento de pacientes fissurados dizem respeito à:
- Alimentação
- Audição
- Cirurgia do lábio
- Cirurgia do palato
- Desenvolvimento da fala
- Rinoplastia
- Enxerto ósseo alveolar
- Cirurgia Ortognática
Alimentação:
As crianças nascidas com uma fissura de palato apresentam uma comunicação da boca com o nariz e não conseguem criar sucção oral. Com isso, vão apresentar dificuldade em conseguir uma quantidade suficiente de leite (ou complemento). As mamadas são naturalmente mais longas e o bebê fica cansado com mais facilidade. É comum o bebê dormir de cansaço e acordar cerca de uma hora depois chorando de fome, o que torna o processo de amamentação frustrante e cansativo para os pais. Ainda assim, o aleitamento materno deve ser estimulado como a melhor opção. Caso o leite materno seja retirado e oferecido em mamadeiras, medidas que podem ajudar a alimentação do bebê fissurado incluem aumentar o furo do bico da mamadeira e usar garrafas do tipo squeeze, para aumentar o aporte de leite para o bebê numa fase inicial. Posteriormente, são utilizados bicos ortodônticos com o furo para cima, confeccionados tanto para a fissura de lábio quanto para a fissura de palato, que ajudam na amamentação. Placas intraorais confeccionadas sob medida para o bebê ajudam a separar a cavidade oral da nasal, facilitando a deglutição. Um posicionamento mais verticalizado do bebê (posição semissentada) também ajuda a melhorar o fluxo do leite e evita a aspiração.

Audição:
Quando a musculatura do palato está normal, a movimentação do palato promove a dilatação e drenagem das tubas auditivas para a rinofaringe. Quando há uma fenda do palato, a sua musculatura se insere de forma incorreta sobre a fenda e não há movimentação adequada das tubas auditivas, propiciando o acúmulo de líquido (otite serosa). O acúmulo crônico pode ocasionar infecções de repetição, o que pode levar, por sua vez, a perdas auditivas. A diminuição da audição em crianças prejudica o desenvolvimento da fala, pois é importante que a criança ouça bem para falar bem e aprender. Com os mecanismos de aprendizado e de fala comprometidos, a criança pode apresentar problemas de comportamento e até alterações psicossociais. Quando a criança está sob risco de perda de audição, pode ser necessária a inserção de um tubo de ventilação (“carretel”), para permitir a drenagem do líquido e aliviar os sintomas. O ideal é que a criança seja avaliada por um Otorrinolaringologista, fazendo audiometrias e avaliando regularmente a sua capacidade auditiva. Caso seja necessária a inserção de tubos de ventilação (“carretel”), isso deve ser coordenado com as cirurgias para o lábio e o palato, de modo a reduzir o número total de cirurgias às quais a criança será submetida.
Esquema da colocação de tubos de ventilação
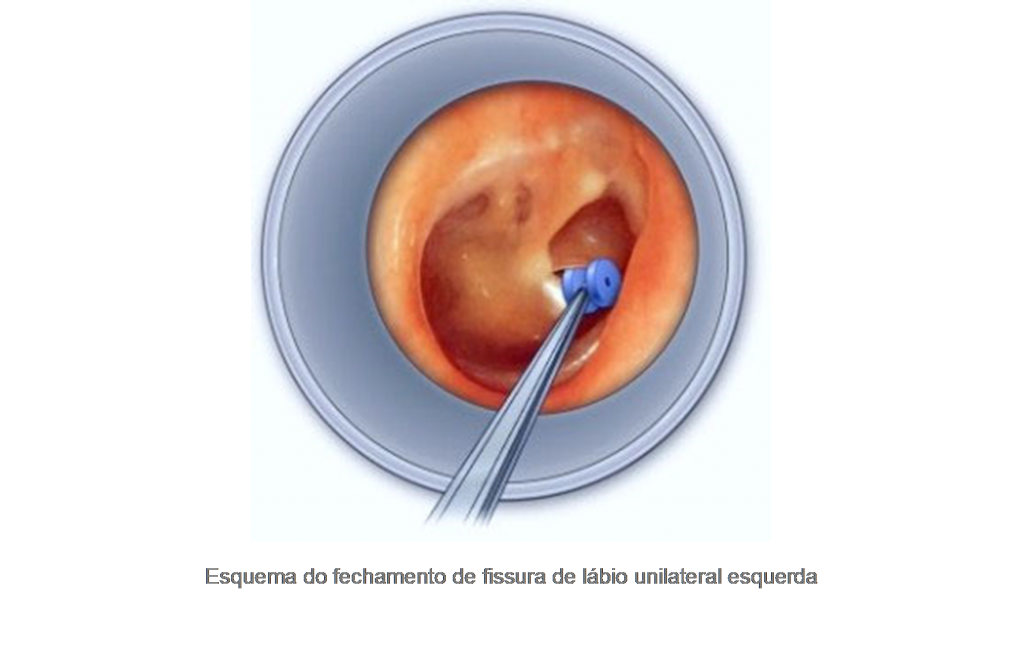
Apesar da ansiedade extrema dos pais, a cirurgia para correção do lábio fissurado não costuma ser realizada em bebês recém-nascidos. Embora alguns trabalhos mostrem a realização da cirurgia em recém-nascidos, sua vantagem está mais relacionada ao tratamento da ansiedade e frustração dos pais que um benefício real para o bebê. Existem algumas regras básicas que devem ser observadas para que a criança possa ser submetida à cirurgia com segurança, principalmente no que diz respeito à sua idade e peso, levando a cirurgia a ser realizada em torno dos 3 meses de idade.
No pré-operatório, embora seja controverso, costumamos fazer uso de uma fita para aproximar as vertentes labiais, diminuindo assim o defeito e facilitando a cirurgia.
Entre as inúmeras técnicas cirúrgicas descritas na literatura, as mais comuns envolvem uma “rotação e avanço” dos tecidos e a confecção de “retalhos triangulares”. A tendência mundial, no entanto, envolve uma flexibilização dos protocolos, com a associação de técnicas que são escolhidas especialmente para cada caso.
Independente da técnica utilizada, alguns objetivos básicos devem ser alcançados em uma cirurgia para correção da fissura labial: reconstrução do fundo de vestíbulo oral (o sulco entre a gengiva e o lábio), reconstrução do assoalho nasal, isolando a cavidade oral da cavidade nasal, reconstrução da cinta muscular labial, permitindo uma adequada movimentação dos lábios, e finalmente, reconstrução da pele e do vermelhão dos lábios, restabelecendo a anatomia normal e a harmonia estética da região.
Apert
Você já ouviu falar da Síndrome de Apert? Apesar de ser pouco conhecida, essa síndrome genética atinge um a cada 100 a 160 mil nascidos vivos.
O que é a Síndrome de Apert?
A síndrome de Apert é uma malformação congênita grave, que associa uma união precoce dos ossos do crânio e da base do crânio (craniofacioestenose).
Essa síndrome é responsável, portanto, por causar o desenvolvimento anormal da estrutura craniana e facial de uma pessoa. Por isso, os bebês portadores dessa malformação nascem com o formato da cabeça e a face diferentes.
A Síndrome de Apert só acontece naqueles que possuem uma mutação em um dos genes responsáveis pelos fibroblastos, que são encarregados de promover a união dos ossos do crânio ainda durante o desenvolvimento do bebê no útero da mãe. Com isso, há uma união precoce e errônea dos ossos da calota craniana e da base do crânio, o que pode prejudicar a estética do crânio, da face e também o desenvolvimento do cérebro da criança.
Além disso, os bebês portadores de Apert também nascem com junções dos dedos das mãos e dos pés que parecem estar todos “colados”, o que também é conhecido como sindactilia complexa. Essa sindactilia de múltiplos dedos é uma característica definidora da síndrome e ajuda a fazer o diagnóstico e a diferenciar de outras síndromes de craniofacioestenoses, como a Síndrome de Crouzon e a Síndrome de Pfeiffer.
Quais são os sintomas da síndrome?
Devido à junção prematura dos ossos do crânio, o cérebro fica muito constrito e pode ter o seu crescimento e desenvolvimento prejudicados. A junção dos ossos da base do crânio, por outro lado, impede que a face cresça normalmente. Com isso, os portadores da Síndrome de Apert terão algumas características físicas reconhecíveis, são elas:
- Face afundada ou retraída;
- Olhos distantes um do outro, devido ao crescimento anormal do crânio;
- Uma testa longa e, consequentemente, uma cabeça de formato mais longo do que o normal.
Além disso, é possível que o portador da síndrome possua os seguintes sintomas: apneia do sono (distúrbio em que a pessoa pára de respirar por alguns segundos enquanto está dormindo), problemas de audição e sucessivas infecções no ouvido e crises constantes de sinusite.
Causas da Síndrome de Apert
Existem dois motivos que podem levar ao desenvolvimento da Síndrome de Apert:
- Um dos progenitores possui o gene: Nesse caso, um dos pais possui em seu DNA o gene que causa a síndrome. Dessa forma, como o bebê carrega metade dos genes do pai e a outra metade da mãe, esse pode ser um dos motivos que leva ao desenvolvimento dessa malformação.
- Mutação genética desconhecida: Assim como diz o nome, pode acontecer casos em que nenhum dos pais possuem a carga genética da síndrome, mas, mesmo assim, acontece uma mutação durante a gestação e o bebê desenvolve o Apert. Mesmo após mais de um século de sua descoberta, ainda não foi possível saber qual é a razão da mutação. Entretanto, alguns estudiosos sobre o assunto afirmam que gestação em idade avançada pode desencadear esse acidente genético.
Devido às suas características físicas, o diagnóstico da Síndrome de Apert pode ser feito logo após o nascimento do bebê ou também a partir de exames genéticos para confirmar a existência da mutação.
Como tratar a síndrome de Apert?
Apesar de não existir uma cura para a síndrome, é possível tratar essa malformação com cirurgias plásticas. Esses procedimentos cirúrgicos são responsáveis por corrigir as alterações no crânio do bebê e os casos de sindactilia. Além disso, a cirurgia permite que o cérebro do recém-nascido consiga se desenvolver e evita que aconteça algum dano mental.
Para melhor eficácia do tratamento, as cirurgias para correção dos sinais craniofaciais da Síndrome de Apert são divididas em três partes:
- Cirurgia para remodelamento craniano: feita nos primeiros meses de vida da criança, essa primeira cirurgia tem como objetivo separar as partes que foram fundidas precocemente no crânio, como uma tentativa de expandir o espaço intracraniano e permitir que o cérebro cresça e se desenvolva normalmente.
- Cirurgia para avanço da face: feita em torno dos 6-8 anos de idade, a cirurgia consiste em “corrigir” os ossos da face, visto que, conforme a criança se desenvolve, a face se mantém retraída e não cresce normalmente. Com isso, o cirurgião responsável tentará colocar os ossos da região do nariz e maxila na posição em que deveriam estar.
- Cirurgia para correção da distância aumentada entre os olhos (hipertelorismo): a terceira e última cirurgia é feita para corrigir a região dos olhos, diminuindo o espaço entre eles.
É importante saber que, apesar de o tratamento craniofacial ser dividido em três cirurgias, isso não significa que não será preciso outras operações durante todo o processo, visto que alguns casos podem ser mais graves do que outros.
Em relação ao tratamento das sindactilias das mãos e pés, é possível realizar a separação dos 10 dedos das mãos e dos pés na grande maioria dos casos. Essa parte do tratamento pode ser dividida em duas etapas cirúrgicas, que podem ser iniciadas ainda no primeiro ano de vida.
Além dos procedimentos cirúrgicos, outro jeito de tratar a Síndrome de Apert inclui tratamentos para melhorar a condição de vida do portador, por exemplo: medicação para tratar sinusites e otites, tratamento intensivo para apneia do sono e utilização de colírios diariamente para prevenir secura nos olhos, entre outros tratamentos que melhorem os sintomas da síndrome.
Dúvidas sobre a Síndrome de Apert
- Qual é a expectativa de vida para os portadores da Síndrome de Apert: A expectativa de vida para uma pessoa que nasceu com a síndrome pode variar. Tudo depende do quanto a anomalia afetou o desenvolvimento do bebê. Por exemplo, uma criança que conseguiu sobreviver aos primeiros anos de vida e não teve mais nenhum tipo de complicação, pode ter uma expectativa de vida igual à de uma que nasceu sem Apert.
- Se eu tiver essa síndrome, meu filho também terá? Por ser uma síndrome genética, caso um dos progenitores tiver o gene, existe 50% de chances do seu filho também nascer com essa malformação craniofacial.
- Caso eu tenha, existe alguma forma de evitar que meu filho nasça com a síndrome? Se você ou seu parceiro (a) apresentam o gene da síndrome, o mais indicado é que utilizar o método de fertilização in vitro para gerar um bebê. Dessa forma, será possível selecionar o embrião que não possui nenhum sinal de Apert. Entretanto, é importante saber que, mesmo que o embrião inicialmente não tenha o gene, é possível que a mutação se desenvolva durante a gestação.
- Meu filho terá um desenvolvimento mental normal? Para que portadores da Síndrome de Apert tenham um desenvolvimento normal, é preciso que a cirurgia de tratamento craniano seja feita o mais rápido possível para evitar que o pouco espaço ou a malformação afetem o crescimento do cérebro. Com as cirurgias corretas e com acompanhamento médico, é muito provável que o bebê consiga se desenvolver normalmente.

Crouzon
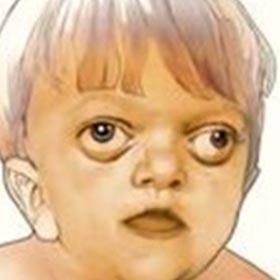
Síndrome descrita por Crouzon em 1912, ela associa uma cranioestenose e uma hipoplasia do terço médio facial. A cranioestenose, na maioria dos casos, acomete as duas suturas coronais.
A alteração facial é característica: hiperteleorbitismo (afastamento das órbitas), exorbitismo (com órbitas rasas, provocando o efeito de “olhos saltados”) e retrusão de todo o terço médio facial. Habitualmente pouco marcante ao nascimento, ela tende a se tornar mais aparente em torno da idade de 2 anos e se agrava progressivamente.
Pfifer
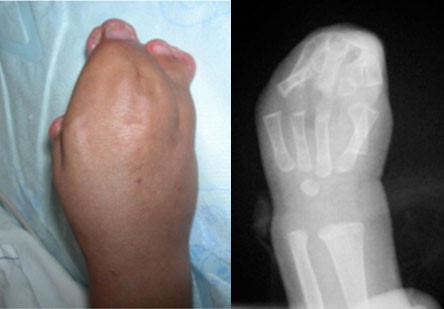
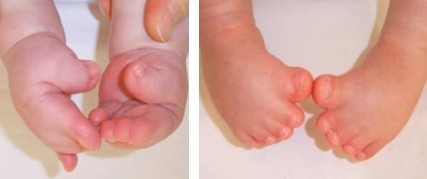
Síndrome descrita por Rudolf Pfeiffer em 1964, ela apresenta uma braquicefalia (fechamento das suturas cranianas coronais) associada a sindactilias membranosas variáveis das mãos e pés e sobretudo um alargamento dos polegares e dos dedões dos pés (“dedão do pé aumentado”), com um desvio marcante para dentro. Uma braquidactilia, uma estenose dos cotovelos, e até mesmo sinfalangia são frequentemente descritos.
Mãos e pés na Síndrome de Pfeiffer, com sindactilias membranosas variáveis e o aumento e o desvio característico dos polegares e dedões dos pés
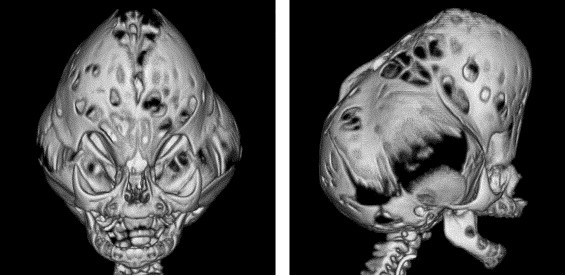
As formas graves, com o crânio em trevo, são descritas. Nestes casos, a dismorfia craniana é muito importante, devido a um bombeamento considerável das fossas temporais e de uma estenose lateral das regiões frontoparietais, levando a um aspecto trilobado visto de frente. A hidrocefalia congênita é constante. Essa dismorfia maior, muitas vezes chamada síndrome do crânio em trevo, pode igualmente ser observada nas síndromes de Apert e na forma precoce da síndrome de Crouzon.
Crânio com alteração característica em formato de trevo
A Síndrome de Pfeiffer é rara, com uma incidência aproximada de 1 para cada 100.000 indivíduos. Ocorre cranioestenoses em associação com polegares e dedões dos pés curtos e desviados para dentro. A fusão prematura das suturas coronais e lambdóides pode ser acompanhada ocasionalmente da fusão ainda da sutura sagital, levando à forma anormal do crânio. Ocorre uma aparência facial característica, com alargamento da cabeça e achatamento do occipital, alongamento da região frontal, hipoplasia (menor desenvolvimento) do terço médio da face, nariz curto com dorso nasal baixo e afastamento dos olhos (hipertelorismo ocular). Os pacientes geralmente apresentam olhos proeminentes (proptose ocular) devido às órbitas serem muito rasas.
Os polegares e os dedões dos pés são curtos e tortos, com um desvio típico. Ocorre junção (sindactilia) do segundo e terceiro dedos. Anomalias adicionais podem incluir: retardo mental em graus variados, estenose de aqueduto cerebral com consequente hidrocefalia, implantação baixa das orelhas, estenose (estreitamento) do conduto auditivo externo, infecções de ouvido recorrentes, e menos frequentemente, hidronefrose, rins pélvicos, bexiga pouco desenvolvida (hipoplásica).
Pacientes com Síndrome de Pfeiffer podem apresentar obstrução de vias aéreas superiores devido à retrusão do terço médio da face.
De acordo com as características clínicas, a Síndrome de Pfeiffer pode ser dividida em 3 subtipos:
Tipo 1: Síndrome de Pfeiffer “clássica”, com manifestações leves incluindo braquicefalia, hipoplasia do terço médio da face e anomalias de dedos das mãos e dos pés. Está associada com desenvolvimento neurológico e intelectual normais e geralmente tem um bom prognóstico.
Tipo 2: caracteriza-se pela deformidade do crânio trilobado, com uma proeminência no topo e uma em cada lado da cabeça, dando um aspecto em forma de “ folha de trevo” (também chamado de Kleeblattschädel), embora essa característica não seja exclusiva da Síndrome de Pfeiffer, podendo ocorrer em outras síndromes ou mesmo isoladamente. Apresenta ainda proptose (olhos saltados) grave, anomalias de dedos das mãos e pés, anquilose (junção) ou estenose de cotovelo, atraso de desenvolvimento mental e complicações neurológicas. O crânio em forma de trevo pode causar limitação ao crescimento cerebral e a proptose grave pode causar perdas visuais importantes.
Tipo 3: semelhante ao tipo 2, porém sem deformidade do crânio em forma de trevo. A ausência do crânio em forma de trevo pode tornar difícil estabelecer o diagnóstico. Os tipos 2 e 3 ocorrem esporadicamente e apresentam um risco aumentado de morte precoce devido ao comprometimento neurológico grave e problemas respiratórios. Uma superposição clínica entre os três tipos pode ocorrer.
Diagnosticando a Síndrome de Pfeiffer
O diagnóstico é geralmente clínico, baseado na presença de cranioestenoses e polegares e dedões dos pés alterados. Geneticamente, ocorrem mutações nos genes dos receptores dos fatores de crescimento dos fibroblastos (do inglês: fibroblast growth factor receptor – FGFR1 e FGFR2). Esses genes atuam na sinalização das células em resposta ao seu ambiente, regulando processos de proliferação, diferenciação e migração celular. Uma mutação em um desses genes causa uma sinalização prolongada, que pode provocar a maturação precoce das células ósseas e fusão prematura dos ossos do crânio, das mãos e dos pés.
Faz diagnóstico diferencial com outras síndromes de craniofacioestenoses, como as Síndromes de Crouzon e Apert, discutidas em outro tópico. De fato, as semelhanças clínicas são grandes, principalmente no que diz respeito ao aspecto orbitário (órbitas rasas com proptose ocular) e à retrusão do terço médio da face, embora geneticamente sejam distintas.
Hipertelorismo Orbitário
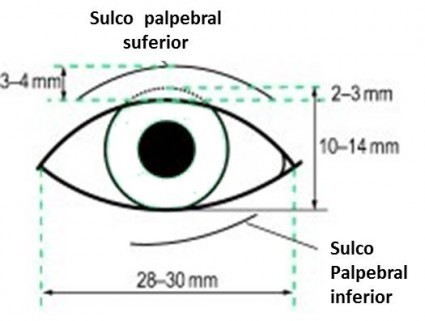
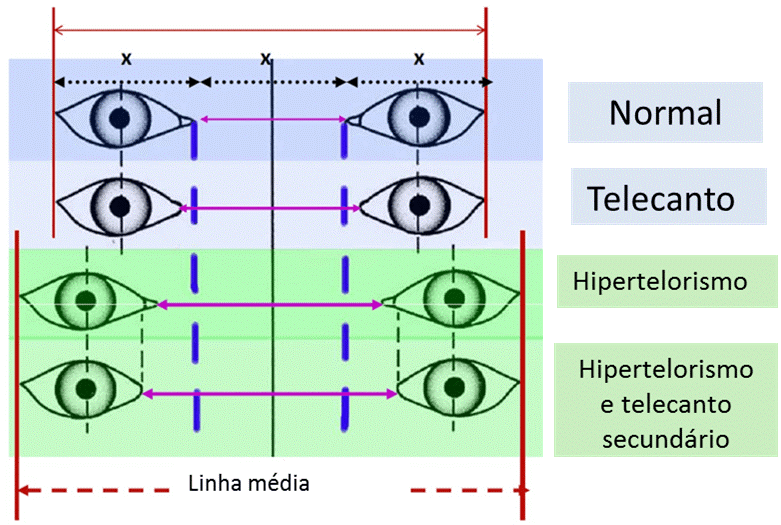
O termo hipertelorismo orbitário ou hiperteleorbitismo ou simplesmente hipertelorismo, corresponde ao aumento da distância entre as órbitas, ou distância interorbital aumentada. A distância entre as órbitas pode ser obtida medindo-se a distância entre os dacrions, ou o ponto onde as suturas frontal, nasal e lacrimal convergem sobre a crista lacrimal anterior, ou seja, a distância entre as margens orbitárias mediais ou internas.
Pode ocorrer um aumento da distância entre os cantos mediais dos olhos sem hipertelorismo verdadeiro, como acontece nos traumas de face em que ocorre telecanto traumático. A distância entre as pupilas também não significa hipertelorismo, uma vez que no estrabismo divergente a distância interpupilar está aumentada, sem haver aumento da distância entre as órbitas.
A distância normal entre as órbitas varia de acordo com a idade do paciente, a raça e o gênero masculino ou feminino. Em geral, toma-se como medida padrão no adulto a distância interorbital de 30mm.
Classificação do Hipertelorismo
O hipertelorismo pode ser classificado em leve, moderado ou grave conforme essa distância entre as órbitas vai aumentando:
Leve: entre 30 e 34mm
Moderado: >34 e <40mm
Grave: acima de 40mm
Causas de hipertelorismo
O hipertelorismo orbitário está relacionado a malformações congênitas, seja por fatores genéticos ou extrínsecos. As principais causas de hipertelorismo são: fissuras raras de face, como a fissura 14 de Tessier, displasia frontonasal, displasia craniofrontonasal (componente genético), encefaloceles frontais, naso-etmoidais, síndromes de Apert e Crouzon.
Alguns problemas estéticos podem acompanhar o hipertelorismo, como encurtamento do nariz, fissura alar, ponta nasal bífida, fronte malformada, implantação anormal da linha do cabelo, afastamento das sobrancelhas, formato anormal dos olhos, mal posicionamento da órbita, distopia do canto lateral dos olhos, encurtamento ou hipoplasia da bochecha, retrusão ou hipoplasia da maxila, obstrução ou alargamento do canal lacrimal, hipertrofia do subcutâneo da área orbito nasal.
